1. Introduction¶
This is an introductory tutorial for learning to work reproducible in bioinformatics/genomics research. This tutorial makes extensive use of the command-line interface.
We will be analysing RNA-seq/transcriptomics samples from yeast. However, in the context of this tutorial the biological background or relevance is of no real importance to us. The aim here is not to understand the data, or bioinformatics tools for that matter, but rather how to structure the analysis steps in a way that you or someone else can redo the analysis and derive at the same results. Yes, the bioinformatics tools we are using here are real and the analyses performed here can be applied to other datasets too, however, all tools can be substituted for alternatives.
Note
The focus in this tutorial is not on the bioinformatics tools, but rather on the tools that facilitate reproducible analyses.
1.1. Prerequisites¶
- This tutorial generally assumes you work in a Unix/Linux type of environment (a minimal tutorial can be assessed here), however, MacOS is fine too.
- You should be relatively comfortable using the command-line interface (you can update your knowledge using the excellent introductory material of the Software Carpentry: here).
- You should have a basic knowledge of version control. We are going to use Git here. A good tutorial, again, is available from the Software Carpentry: here
- Any bioinformatics knowledge is a bonus but not strictly required (a Genomics tutorial can be assessed here).
1.2. Learning outcomes¶
During this tutorial you will learn to:
- Use conda and in particular Bioconda [GRUENING2018] for tool installation and tracking of the version numbers of the used tools.
- Use Snakemake [KOESTER2012] to create workflows / pipelines to analyse your data in a way that is accessible and reproducible.
- Understand how containerization of software can facilitate reproducibility in an operating system independent manner.
- Use Git as a means to version control our analysis workflow and make it publicly available.
1.3. The data we will be using¶
In this tutorial we will analyse public data stemming from a transcriptomics experiment (RNA-sequencing) using next-generation sequencing (NGS). The associated publication is entitled “Dynamics of the Saccharomyces cerevisiae Transcriptome during Bread Dough Fermentation” and can be found here [ASLANKOOHI2013]. The associated data has been deposited at the Short Read Archive and can be found here (accession: PRJNA212389). The final aim in this tutorial is to quantify the expression of genes in each sample.
Note
The data has been downloaded already and is being made available within a Git repository accompanying this tutorial. To facilitate timely analyses during this tutorial, the original data has been down-sampled.
An overview of a typical RNA-seq experiment can be seen in Fig. 1.1. RNA gets extracted from samples of interest and reverse transcribed and sequenced as a proxy for gene expression of the sample (either a set of cells or single cell).
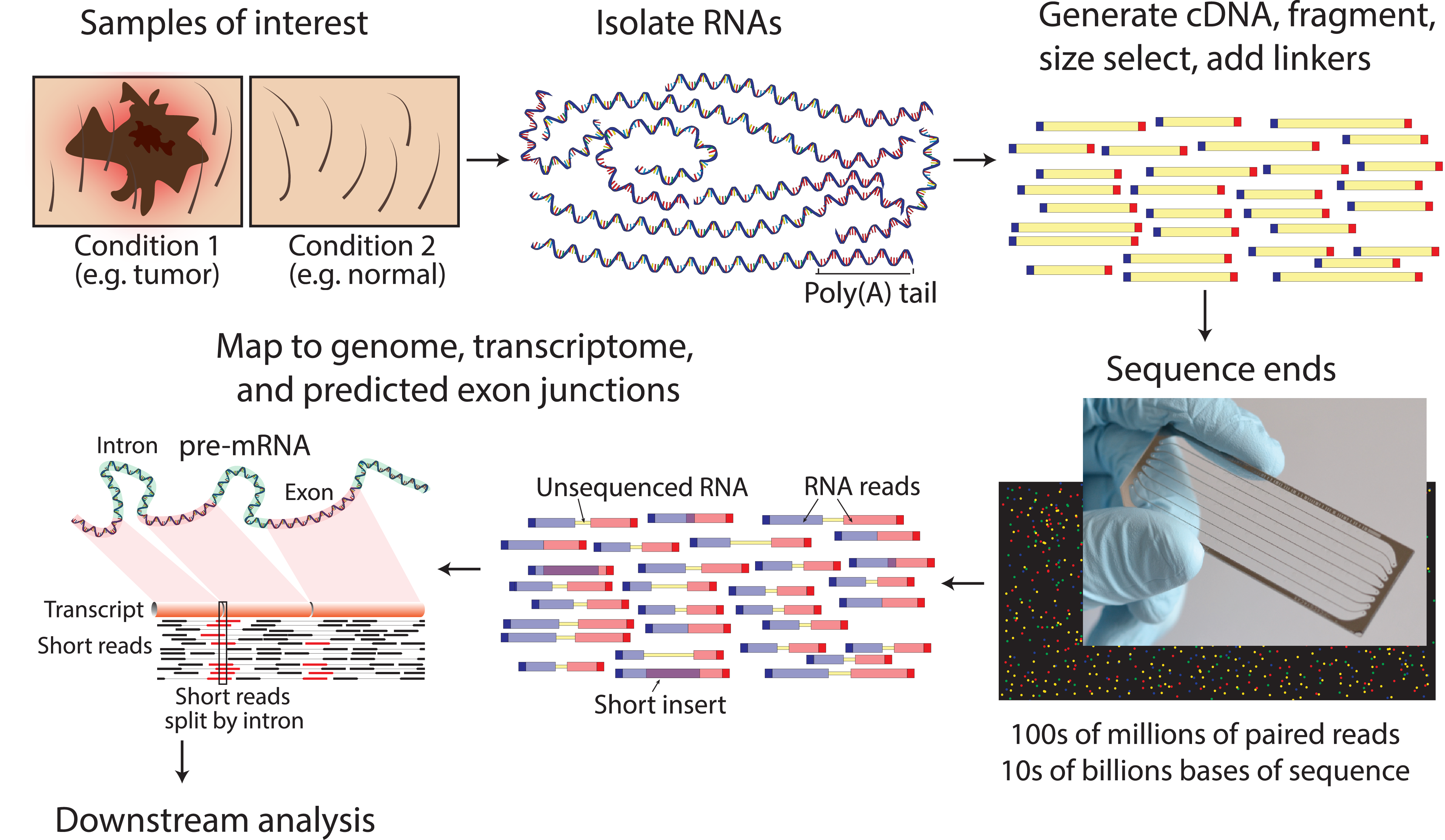
Fig. 1.1 RNA-seq overview (from https://doi.org/10.1371/journal.pcbi.1004393) [GRIFFITH2015].
1.4. The analysis workflow¶
We will be using a traditional set-up for analysing RNA-seq data, where sequenced reads will be cleaned, mapped to a reference genome, and finally reads per transcript/gene-model counted. The workflow is summarised in Fig. 1.2.
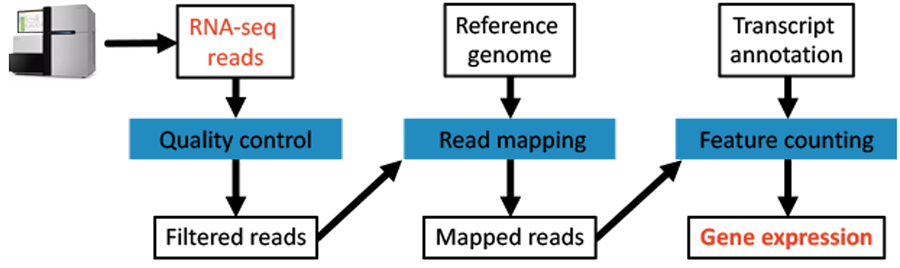
Fig. 1.2 The tutorial will analyse data using this workflow.
- Quality control - We will be filtering reads based on read quality.
- Read mapping - We Will be using a tool for mapping reads to the human genome.
- Feature counting - We will count reads per gene model to derive gene expression values.
References
| [GRUENING2018] | Grüning B, et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nature Methods, 2018, 15:475–476. |
| [KOESTER2012] | Köster J and Rahmann S. Snakemake - A scalable bioinformatics workflow engine. Bioinformatics 2012, 10.1093/bioinformatics/bts480. |
| [ASLANKOOHI2013] | Aslankoohi E, et al. Dynamics of the Saccharomyces cerevisiae Transcriptome during Bread Dough Fermentation. Appl Environ Microbiol. 2013 Dec; 79(23): 7325–7333. |
| [GRIFFITH2015] | Griffith M, Walker JR, Spies NC, Ainscough BJ, Griffith OL. Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud. PLoS Comput Biol. 2015 Aug 6;11(8):e1004393. doi: 10.1371/journal.pcbi.1004393. |