4. Creating analysis workflows¶
4.1. What is a workflow management system?¶
A workflow management system provides an infrastructure for the setting up, performing and monitoring defined sequences of commands, hence addressing our second requirement:
Keeping track of the commands used to analyse the data, including tool parameters.
There are many such systems and some have been specifically designed for genomics and bioinformatics tasks. An recent overview can be accessed here [LEIPZIG2017].
4.2. What is Snakemake?¶
Snakemake is a workflow engine, developed for creating scalable bioinformatics and genomics workflows [KOESTER2012]. It borrows ideas from an old system for compiling and link a program Make and extends the ideas to help with bioinformatics pipelines. We will be using Snakemake to run our complete analysis for us, from start to end. Snakemake will make sure that our jobs are run in the correct order and will recognise if jobs have already been run and thus do not have to be run again. It will also recognise if input files changed and thus jobs have to be re-run. Correctly configured, Snakemake will take care of error logging, benchmarking, and simultaneous execution of our jobs (it is also able to distribute jobs to computer clusters). We will see that combined with conda it makes for a powerful system for developing and running reproducible analyses workflows.
4.3. Setup¶
4.3.1. Install Snakemake¶
We are going to create a general conda environment and install Snakemake into it.
$ conda create -n tutorial python=3 snakemake
# activate the environment
$ source activate tutorial
4.3.2. Download data¶
I prepared a Git repository that contains the some data we will work with. You can download the repository using:
$ git clone https://gitlab.com/schmeierlab/reproduce-tutorial.git
# or try
$ git clone [email protected]:schmeierlab/reproduce-tutorial.git
# Change into the created directory
$ cd reproduce-tutorial
# Let delete the associated git remote
$ git remote remove origin
Note
If the cloning with Git does not work, you can download a zipped
archive of the whole repository here
https://gitlab.com/schmeierlab/reproduce-tutorial/-/archive/master/reproduce-tutorial-master.zip. The
locally on the command-line, you can type unzip
reproduce-tutorial-master.zip.
Let’s investigate the directory:
$ tree
.
├── README.md
├── Snakefile
├── analyses
│ └── results
│ └── README.md
├── data
│ ├── Saccharomyces_cerevisiae.R64-1-1.92.gtf.gz
│ └── Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa
├── envs
│ ├── map.yaml
│ ├── sickle.yaml
│ ├── snakemake-kubernetes.yaml
│ ├── snakemake.yaml
│ └── subread.yaml
├── examples
│ ├── Snakefile_v2
│ ├── ...
├── fastq
│ ├── SRR941826.fastq.gz
│ ├── SRR941827.fastq.gz
│ ├── SRR941830.fastq.gz
│ └── SRR941831.fastq.gz
└── help
├── bwa.help
├── featureCounts.help
├── sickle.se.help
└── snakemake.help
This directory contains all the files we need to do this tutorial.
There are four fastq-files in the fastq directory that we want to clean and map to the reference genome.
Finally, we will count the reads per gene and per sample.
The complete workflow is depicted in Fig. 4.1.
4.4. The analysis without a workflow management system¶
We can of course do this analysis without any workflow management system and write down the commands one by one.
Given that we only have four samples, this is not particular difficult.
However, this process does not scale well if we decide to do it for 400 samples.
So we are later going to use a workflow management system that creates for us the commands for each sample without us doing it explicitly.
However, here we are going to look at each step and write down the command for one of the samples (SRR941826) to understand what is involved.
Activate our conda environment for the tutorial session:
conda activate tutorial
4.4.1. Data QC¶
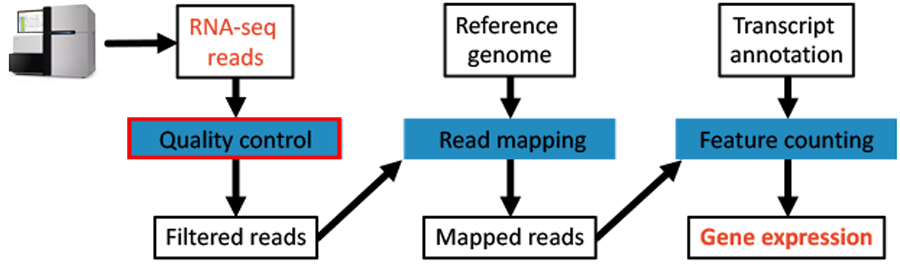
Fig. 4.1 The workflow: Data QC step.
The purpose of this step is to remove bases from the ends of the reads that are of bad quality. There are many tools that can do this for us. Here, we are going to use the program Sickle to perform this task [JOSHI2011]. The program is easy to use.
After installing Sickle, running sickle by itself will print the help:
# install sickle
$ conda install sickle-trim
$ sickle
Running sickle with either the single-end (se) or paired-end (pe) reads:
sickle se
sickle pe
Here the command for our single-end fastq-file:
$ sickle se -g -t sanger -f fastq/SRR941826.fastq.gz -o analyses/results/SRR941826.trimmed.fastq.gz
-g: will facilitate gzip output-t: specifies the quality metric used in the fastq-file-f: input filename-o: output filename
Easy enough! Lets map reads.
4.4.2. Read mapping¶
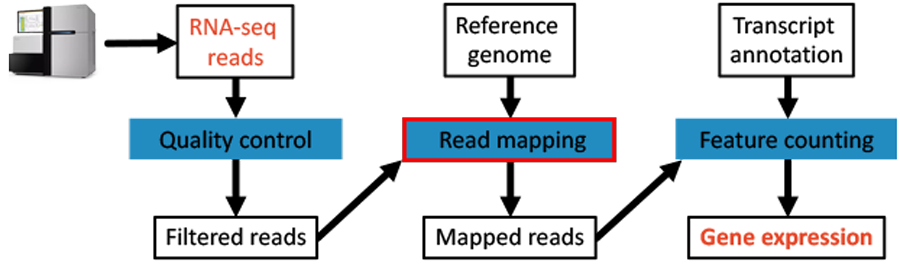
Fig. 4.2 The workflow: The mapping step.
We use BWA and SAMtools to get mapped reads in bam-format. In order to map the reads to the genome we first need to index the genome:
$ conda install bwa samtools
$ bwa index data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa
Now we can take a sample and map it:
$ bwa mem -t 8 data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa analyses/results/SRR941826.trimmed.fastq.gz
| samtools view -Sbh > analyses/results/SRR941826.bam
4.4.3. Feature counting¶
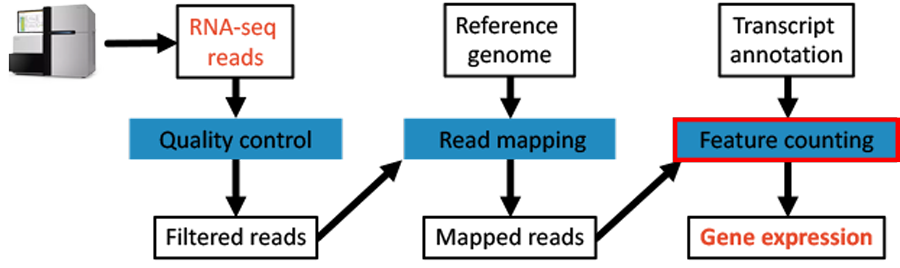
Fig. 4.3 The workflow: Expression quantification step.
We are using the featureCounts tool of the subread package to count reads per feature.
$ conda install subread
$ featureCounts -T 4 -t exon -g gene_id
-a data/Saccharomyces_cerevisiae.R64-1-1.92.gtf.gz
-o counts.txt analyses/results/SRR941826.bam
-T: Number of threads to use at the same time-t: Specify feature type in GTF annotation. Features used for read counting will be extracted from annotation using the provided value.-g: Specify attribute type in GTF annotation-a: The annotation file with the features-o: Output file
4.4.4. Saving tool version information¶
Great, we have done our analysis of all four samples. Now, we can export our conda environment and save the information in a file:
$ conda activate tutorial
$ conda env export > tutorial.yaml
This file contains the tools and their versions that we used in this analysis. We could give this file to someone else and they could, given they work on the same operating system, recreate the same conda environment and redo the analysis.
4.4.5. Summary¶
So, generally we could use the programs on the command-line like shown above with one sample after the other. However, we do not want to do this, as in this manner we do not save the information about the commands we used. We could of course put all commands in a bash-script and in this manner remember all commands that have been run.
However, we would still not be able to keep this approach general to run it again and again on different (numbers) of samples. We can do better! This is were Snakemake comes into play.
4.5. Using a workflow management system¶
Let’s look at the complete workflow again for one samples:
# 1 Trimming
$ sickle se -g -t sanger -f fastq/SRR941826.fastq.gz -o analyses/results/SRR941826.trimmed.fastq.gz
# 2 Genome indexing
$ bwa index data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa
# 3 Read mapping
$ bwa mem -t 8 data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa analyses/results/SRR941826.trimmed.fastq.gz
| samtools view -Sbh > analyses/results/SRR941826.bam
# 4 Counting reads per features
$ featureCounts -T 4 -t exon -g gene_id
-a data/Saccharomyces_cerevisiae.R64-1-1.92.gtf.gz
-o counts.txt analyses/results/SRR941826.bam
We will engineer now this workflow in the workflow management system Snakemake. However, it will be general with no filenames hard-coded, so that we can run the same workflow on any arbitrary number of fastq-files of the same type, here single-end reads.
4.5.1. Snakemake¶
Snakemake uses rules that define how to get from an input to an output.
These rules are defined in a Snakefile that is read upon Snakemake execution.
A basic structure of a rule looks like this:
1 2 3 4 5 6 7 | rule do-something:
input:
"{sample}.fastq"
output:
"{sample}.out"
shell:
"SOMECOMMAND {input} {output}"
|
In this example, we have an input file and an output file, as well as a way to get from the input to the output via a shell command. Rules can either use shell commands, plain Python code or external scripts to create output files from input files. Curly brackets define wildcards that get substituted.
Once, you have written lots of rules, Snakemake determines the rule dependencies by matching file names.
Let us write a rule for trimming to see how this works in practice.
4.5.2. Creating rules¶
Let us build a general rule for the first step, the read trimming via sickle.
The original command was:
$ sickle se -g -t sanger -f fastq/SRR941826.fastq.gz -o analyses/results/SRR941826.trimmed.fastq.gz
We will use this command in our first rule and substitute the input and output part, as well as some of the parameters with wildcards.
In the working directory their is an empty Snakefile.
We will add to this file during the tutorial.
Open this file in a text editor.
1 2 3 4 5 6 7 8 9 | rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
|
The command in the shell keyword section will be used to trim the data.
However, before it is executed, Snakemake will replace the wildcards in the command with the proper values defined in the other sections of the rule.
Of note, we introduce here as well a keyword params (highlighted lines), with which one can add more flexibility to the values that get substituted in the shell command.
Fine, but what is happening with the strange {sample} wildcard?
The wildcard will be replaced by Snakemake to try and match our requested final targets.
However, we have not defined any targets yet.
Snakemake needs to know for what to run this rule. We need to define result or target files we want to create.
Note
1. Snakemake works by matching file-names, i.e. finding rules that can generate the requested files from other files. 2. Snakemake automatically creates directories if they do not already exist (e.g. analyses/results/).
Lets define some targets.
We are creating a pseudorule (all) that only defines inputs, which are our expected final targets.
In this way, Snakemake finds the rule all and tries to figure out which rules can be run to create the desired output target files.
It will find that our rule trimse can accomplish this when run four times with four different input files by substituting the {sample} wildcard.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 | rule all:
input:
["analyses/results/SRR941826.trimmed.fastq.gz",
"analyses/results/SRR941827.trimmed.fastq.gz",
"analyses/results/SRR941830.trimmed.fastq.gz",
"analyses/results/SRR941831.trimmed.fastq.gz"]
rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
|
We defined four target files. Let us look what happens here for one of the target files when you run Snakemake.
- Snakemake finds that you request to create the file
analyses/results/SRR941826.trimmed.fastq.gz. - Snakemake will scan all rules, to identify which rule can create this file (Snakemake will try to substitute any wildcards like
{sample}and try to match file names). - In our case, it will find that substituting
{sample}in the output section of ruletrimse(analyses/results/{sample}.trimmed.fastq.gz) withSRR941826will match the requested target file nameanalyses/results/SRR941826.trimmed.fastq.gz - Snakemake will check if the input file of rule
trimse(fastq/{sample}.fastq.gz) with a substituted{sample}part withSRR941826(fastq/SRR941826.fastq.gz) exists. - If this file can be found the rule will be scheduled for execution. If the input cannot be found, Snakemake will complain that the requred input for creating the requested file is missing.
Ok, this Snakefile can already be run with the snakemake command.
We can do a dry-run (without actually running anything) to see the commands that Snakemake would execute.
We use the snakemake flag -n for dry-run and -p to see the commands that Snakemake would execute:
$ snakemake -np
Building DAG of jobs...
Job counts:
count jobs
1 all
4 trimse
5
rule trimse:
input: fastq/SRR941830.fastq.gz
output: analyses/results/SRR941830.trimmed.fastq.gz
jobid: 3
wildcards: sample=SRR941830
sickle se -g -t sanger -f fastq/SRR941830.fastq.gz -o analyses/results/SRR941830.trimmed.fastq.gz
...
Nice, the Snakemake correctly substituted the files in input and output to create the correct commands for trimming.
Note
We used explicit file names for the expected target files in rule all based on the input sample file names we knew existed in the fastq directory . However, we want to be general to be able to run any files located in the fastq directory without explicitly listing them. Snakemake should identify the input files and create expected target file names automatically based on the input file names.
Lets rewrite it a bit to make the workflow more general, so that any file located in the fastq directory that has the right file name structure is found by Snakemake:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 | SAMPLES, = glob_wildcards("fastq/{sample}.fastq.gz")
rule all:
input:
expand("analyses/results/{sample}.trimmed.fastq.gz", sample=SAMPLES)
rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
|
- We use an inbuilt function
glob_wilcardsto scan thefastqdirectory for files of a certain structure and extract the sample identifier out of the file names. - We use another inbuilt function
expandin ruleallto create a new “target” file name for each sample identifier collected in the step before.
Running the same snakemake -np command again, will yield the same result as in the explicit case.
However, now it would not matter if we would add another 100 files to the fastq directory.
Snakemake would find them, without us doing anything else.
4.5.3. Error logging and benchmarking¶
There are a few things we can still add to our rule to facilitate error logging and benchmarking (the process of testing how long a task takes):
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | SAMPLES, = glob_wildcards("fastq/{sample}.fastq.gz")
rule all:
input:
expand("analyses/results/{sample}.trimmed.fastq.gz", sample=SAMPLES)
rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
log:
"analyses/logs/{sample}.trimse"
benchmark:
"analyses/benchmarks/{sample}.trimse"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
" 2> {log}"
|
- We are adding another keyword (
log), specify the file where we want the logging to end up in (of note: it also contains the same wildcard as the input and output files, thus gets substituted as well), and add the error redirection to the wildcardlogin the shell command. - We add another keyword (
benchmark) and specify the file where we want the data to end up in. We do not do anything else. Snakemake will take care of benchmarking for us.
Note
You should realise that the shell command can be written over several lines, like in the example above.
If you rerun the snakemake command logging and benchmarking will be visible in the execution plan of Snakemake.
Because we specified the same wildcard in the benchmark file name, it gets substituted with the sample identifier too.
$ snakemake -np
Building DAG of jobs...
Job counts:
count jobs
1 all
4 trimse
5
rule trimse:
input: fastq/SRR941830.fastq.gz
output: analyses/results/SRR941830.trimmed.fastq.gz
log: analyses/logs/SRR941830.trimse
jobid: 3
benchmark: analyses/benchmarks/SRR941830.trimse
wildcards: sample=SRR941830
sickle se -g -t sanger -f fastq/SRR941830.fastq.gz -o analyses/results/SRR941830.trimmed.fastq.gz 2> analyses/logs/SRR941830.trimse
...
4.5.4. Integrating package management¶
Now that we are after reproducibility, we need to integrate package management into the workflow. This is easily done with Snakemake using conda. We can add another keyword argument to our rule that specifies the conda environment that will be activated before running the particular rule.
You can find a minimal conda environment file for sickle in the envs directory: sickle.yaml.
1 2 3 4 5 6 | channels:
- bioconda
- conda-forge
- defaults
dependencies:
- sickle-trim ==1.33
|
The file specifies that the rule should be run with sickle version 1.33.
Before running anything, Snakemake will create the environment and store it in the current working directory in a subdirectory of .snakemake.
It will only be recreated if the yaml file changes.
Otherwise, if Snakemake is rerun it will use the already created environment.
Let us integrate conda and the environment in our sickle rule:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 | SAMPLES, = glob_wildcards("fastq/{sample}.fastq.gz")
rule all:
input:
expand("analyses/results/{sample}.trimmed.fastq.gz", sample=SAMPLES)
rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
log:
"analyses/logs/{sample}.trimse"
benchmark:
"analyses/benchmarks/{sample}.trimse"
conda:
"envs/sickle.yaml"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
" 2> {log}"
|
In order to tell the snakemake command that we want to make use of conda environments in our rules, we need to specify the --use-conda flag when running Snakemake.
Note
Integrating conda’s environment capability in Snakemake is a powerful way to keep track of what tools and versions of tools have been used.
4.5.5. Running Snakemake¶
Now that we have our first rules established, let us run Snakemake and look at the outputs.
In order to actually run the Snakemake workflow we need to adjust the snakemake command a bit:
$ snakemake -p --use-conda
We got rid of the -n flag that signalled a dry-run.
We also added the --use-conda flag as we want Snakemake to use conda environments when running our rules.
Let us look at the results in the analyses folder:
$ tree analyses
analyses/
├── benchmarks
│ ├── SRR941826.trimse
│ ├── SRR941827.trimse
│ ├── SRR941830.trimse
│ └── SRR941831.trimse
├── logs
│ ├── SRR941826.trimse
│ ├── SRR941827.trimse
│ ├── SRR941830.trimse
│ └── SRR941831.trimse
└── results
├── SRR941826.trimmed.fastq.gz
├── SRR941827.trimmed.fastq.gz
├── SRR941830.trimmed.fastq.gz
└── SRR941831.trimmed.fastq.gz
3 directories, 12 files
$ cat analyses/benchmarks/SRR941826.trimse
s h:m:s max_rss max_vms max_uss max_pss io_in io_out mean_load
0.7001 0:00:00 4.79 19.03 0.86 0.95 0.00 0.60 0.00
$ cat analyses/logs/SRR941826.trimse
$
Our result files, as well as benchmarks and log files, have been created.
If we would rerun snakemake, it would tell us that there are no jobs needed to run as all requirements (targets) are satisfied.
$ snakemake -p --use-conda
Building DAG of jobs...
Nothing to be done.
Complete log: .../reproduce-tutorial/.snakemake/log/2018-06-12T142827.292869.snakemake.log
Let us delete a particular result file to test if Snakemake will realize that one sample is missing and still needs to be run:
$ rm analyses/results/SRR941826.trimmed.fastq.gz
$ $ snakemake -n --use-conda
Building DAG of jobs...
Job counts:
count jobs
1 all
1 trimse
2
rule trimse:
input: fastq/SRR941826.fastq.gz
output: analyses/results/SRR941826.trimmed.fastq.gz
log: analyses/logs/SRR941826.trimse
jobid: 3
benchmark: analyses/benchmarks/SRR941826.trimse
wildcards: sample=SRR941826
localrule all:
input: analyses/results/SRR941827.trimmed.fastq.gz, analyses/results/SRR941826.trimmed.fastq.gz, analyses/results/SRR941831.trimmed.fastq.gz, analyses/results/SRR941830.trimmed.fastq.gz
jobid: 0
Job counts:
count jobs
1 all
1 trimse
2
Indeed, Snakemake finds that we still need to run the rule trimse once to fulfil all requirements in rule all.
Note
Benchmark and logging files will be overwritten in subsequent runs for the same sample.
4.5.6. Visualising the workflow graph¶
Internally, Snakemake is creating a directed acyclic graph (DAG) of the rules and their dependencies. We can generate a graphical visualisation of the graph and thus workflow (Fig. 4.4) using Snakemake and Graphviz:
$ snakemake --dag | dot -Tpng > dag.png
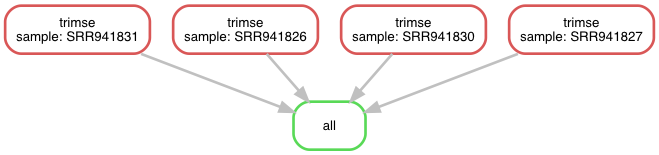
Fig. 4.4 The workflow v5 as a directed acyclic graph.
Note
This visualisation becomes bigger and bigger with more and more samples.
4.5.7. Building the remaining rules¶
We are adding two more rules.
One rule for indexing of the genome using BWA and the another that will take the index, as well as a sample fastq file, and map the reads to the genome.
We are adding to our Snakefile.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 | SAMPLES, = glob_wildcards("fastq/{sample}.fastq.gz")
rule all:
input:
expand("analyses/results/{sample}.bam", sample=SAMPLES)
rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
log:
"analyses/logs/{sample}.trimse"
benchmark:
"analyses/benchmarks/{sample}.trimse"
conda:
"envs/sickle.yaml"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
" 2> {log}"
rule makeidx:
input:
fasta = "data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa"
output:
touch("data/makeidx.done")
log:
"analyses/logs/makeidx.log"
benchmark:
"analyses/benchmarks/makeidx.txt"
conda:
"envs/map.yaml"
shell:
"bwa index {input.fasta} 2> {log}"
rule map:
input:
reads = "analyses/results/{sample}.trimmed.fastq.gz",
idxdone = "data/makeidx.done"
output:
"analyses/results/{sample}.bam"
log:
"analyses/logs/{sample}.map"
benchmark:
"analyses/benchmarks/{sample}.map"
threads: 8
conda:
"envs/map.yaml"
params:
idx = "data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa"
shell:
"bwa mem -t {threads} {params.idx} {input.reads} | "
"samtools view -Sbh > {output} 2> {log}"
|
There are a few points that need a closer look:
- On line 5 we are changing the final output to
bamfiles, so that the mapping is run for all samples. - The shell command to index the genome with BWA does not create an output. Thus, we create a pseudo output. On line 28 We are using the function
touchto create an empty file after the rule is successfully run. We require this file as input for the map rule on line 41 so that the indexing rule is run before any mapping happens. - Line 41 also shows that we can have more than one input to a rule.
- We see a new keyword in a rule on line 48
threads. This can be used to specify the number of threads allowed for the rule when runningsnakemakewith the--jobs NUMflag. - Line 54 and 55 show that we can chain shell commands easily in a Snakemake rule.
- We also added a separate conda environment for indexing and mapping.
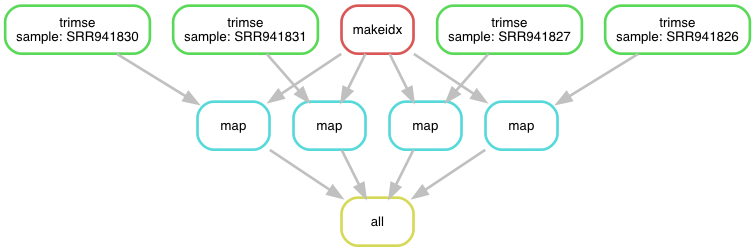
Fig. 4.5 The workflow v6 as a directed acyclic graph.
Finally, we add the rule to count the reads per features.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 | SAMPLES, = glob_wildcards("fastq/{sample}.fastq.gz")
rule all:
input:
"analyses/results/counts.txt"
rule trimse:
input:
"fastq/{sample}.fastq.gz"
output:
"analyses/results/{sample}.trimmed.fastq.gz"
log:
"analyses/logs/{sample}.trimse"
benchmark:
"analyses/benchmarks/{sample}.trimse"
conda:
"envs/sickle.yaml"
params:
qualtype="sanger"
shell:
"sickle se -g -t {params.qualtype} -f {input} -o {output}"
" 2> {log}"
rule makeidx:
input:
fasta = "data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa"
output:
touch("data/makeidx.done")
log:
"analyses/logs/makeidx.log"
benchmark:
"analyses/benchmarks/makeidx.txt"
conda:
"envs/map.yaml"
shell:
"bwa index {input.fasta} 2> {log}"
rule map:
input:
reads = "analyses/results/{sample}.trimmed.fastq.gz",
idxdone = "data/makeidx.done"
output:
"analyses/results/{sample}.bam"
log:
"analyses/logs/{sample}.map"
benchmark:
"analyses/benchmarks/{sample}.map"
threads: 8
conda:
"envs/map.yaml"
params:
idx = "data/Saccharomyces_cerevisiae.R64-1-1.dna_sm.toplevel.fa"
shell:
"bwa mem -t {threads} {params.idx} {input.reads} | "
"samtools view -Sbh > {output} 2> {log}"
rule featurecount:
input:
gtf = "data/Saccharomyces_cerevisiae.R64-1-1.92.gtf.gz",
bams = expand("analyses/results/{sample}.bam", sample=SAMPLES)
output:
"analyses/results/counts.txt"
log:
"analyses/logs/featurecount.log"
benchmark:
"analyses/benchmarks/featurecount.txt"
conda:
"envs/subread.yaml"
threads: 4
shell:
"featureCounts -T {threads} -t exon -g gene_id -a {input.gtf} -o {output} {input.bams}"
" 2> {log}"
|
- We need to change the input of rule
allagain to specify our final target (line 5). - On line 60 we are specifying all
bamfiles as input, asfeatureCountscan be run on all samples at the same time to produce one table of counts for all samples.
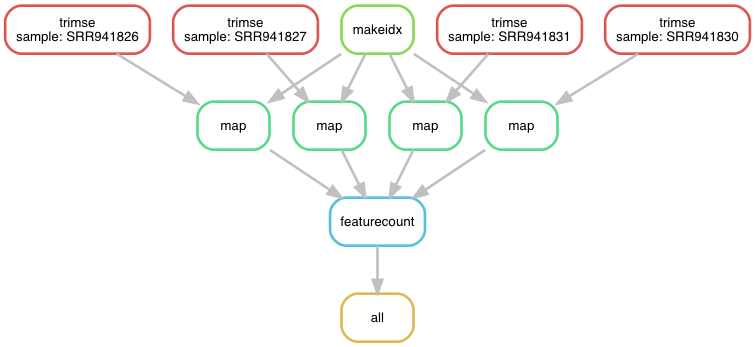
Fig. 4.6 The workflow v7 as a directed acyclic graph.
This looks very similar to our original workflow cartoon from the beginning.
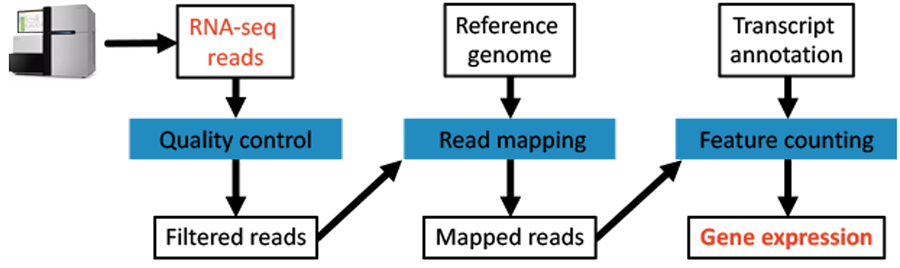
Fig. 4.7 The original workflow.
4.6. Making your work available¶
We are left with one requirement that we wanted to address in this tutorial:
Publishing & versioning the workflow information, as to keep track of when workflows change and what changes occurred.
After we created our workflow and our tool specifications in form of yaml files, we can make sure that others are able to easily get to our workflow specifications.
The easiest way to facilitate this, is to put your directory under Git version control and push your repository to a remote provider like GitLab or GitHub.
The URL to this repository can be added to manuscripts or sent via emails to collaborators so that others can easily download the repository and thus redo the analysis with the same settings, hence we are fulfilling our last requirement above.
$ git init
$ git add Snakefile
$ git add envs/*
# commit your changes
$ git commit -m "Init"
# Create a remote on GitLab, GitHub, Bitbucket, etc.
# and associate remote to repository
$ git remote add origin [email protected]/...
$ git push -u origin master
Note
It might be tempting to add the input data as well to the repository. However, in most cases it will be very big and you are better of using a remote storage location for the input data, e.g. Dropbox, Google Cloud Storage, FTP, etc. Snakemake can deal with all of those remote locations and can download data on demand. An example can be seen in the example file “Snakemake_v8” (see also below), where I request the input data from a Google Cloud Storage bucket. Of course there might be fees associated with storing data in the cloud. For proper reproducibility it should be a persistent location. For genomic data, free storage solutions like NCBI Short Read Archive or Gene Expression Ominbus can be used.
1 2 3 4 5 6 7 8 9 10 11 12 | from snakemake.remote.GS import RemoteProvider as GSRemoteProvider
GS = GSRemoteProvider()
# Read samples from a Google Cloud Storage bucket.
# Samples will be downloaded, processed locally and uploaded to google storage
# Call snakemake with the --default-remote-provider GS --default-remote-prefix BUCKET-PREFIX
SAMPLES, = GS.glob_wildcards("schmeier-reproduce-bucket/fastq/{sample}.fastq.gz")
rule all:
input:
expand("analyses/results/{sample}.trimmed.fastq.gz", sample=SAMPLES)
...
|
References
| [LEIPZIG2017] | Leipzig J. A review of bioinformatic pipeline frameworks. Briefings in Bioinformatics, Volume 18, Issue 3, 1 May 2017, Pages 530–536, |
| [JOSHI2011] | Joshi NA, Fass JN. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. (2011) Available at https://github.com/najoshi/sickle. |